Contents
- Project Info
- Database Accounts
- Database Model and Privileges
- How do you model a scientific workflow within the database?
- How are access privileges granted with respect to this model?
- Biological Samples
- What kinds of samples does the database accept, and what attributes are stored?
- How do I submit my sample(s) to the database?
- What is a barcode file?
- What is a samplesheet?
- Assays and Datasets
- Accessing Your Data
- How do I know when my data is ready?
- Who can see my data?
- How do I search for my data later on?
- How do I download my data?
- How do I upload my data to Galaxy?
- How can I share my data with external collaborators?
- Miscellaneous
- What's the relationship between an iLab account, a Princeton account, and an HTSEQ account?
- What's the difference between a sample's name and its tube identifier?
- Why are the fastq filenames so long and how can I make them shorter?
- How do I delete my data from the database?
- How do I run Cell Ranger on my data?
- How can I find out more about this project?
- Project Info
- What is this project?
- Database Accounts
- How do I acquire an account?
- Princeton University researchers, OR
- their sponsored external collaborators, OR
- sponsored customers of the Princeton University Genomics Core
- I've forgotten my username and password, and now I can't get in. Could you check on this for me?
- I can't log in?
- Make sure that the Caps Lock key is not depressed. Login names are not case sensitive, but passwords are.
- Make sure your browser is set to accept cookies.
- Database Model and Privileges
- How do you model a scientific workflow within the database?
- How are access privileges granted with respect to this model?
- Biological Samples
- What kinds of samples does the database accept, and what attributes are stored?
- How do I submit my sample(s) to the database?
- individual submission, via webform
- batch submission, via either webform or file upload
- What is a barcode file?
- What is a samplesheet?
- Assays and Datasets
- What is an assay?
- What is a dataset?
- What is the experimental graph?
- Retrieving Your Data
- How do I know when my data is ready?
- Who can see my data?
- How do I search for my data later on?
- Use the "Search" box at the top of each page. This returns your data in the form of graphs. For
best results use a
single word or string (search does not yet support boolean operators). There is more information on how the
search is performed here. - Use the "My Data" menu items which let you view your samples, assays and datasets in tabular form, as well as your graphs.
- Use the "Search" menu items, which have options for searching for samples, assays and tags. The
data is displayed in
tabular form. - How do I download my data?
- How do I upload my data to Galaxy?
- How can I share my data with external collaborators?
- Miscellaneous
- What's the relationship between an iLab account, a Princeton account, and a HTSEQ account?
- What's the difference between a sample's name and its tube identifier?
- Why are the fastq filenames so long and how can I make them shorter?
- How do I delete my data in HTSEQ?
- How do I run Cell Ranger on my data?
- How can I find out more about this project?
The HTSEQ project tracks sample data, protocol and analysis parameters, and provides standardized data and reports for high throughput sequencing data. Our project is fairly tightly coupled with the local Genomics Core Facility and is integrated with their utilization of the University's iLab Portal, primarily importing and processing the Core-handled samples and jobs. After sequencing by the Core (Illumina platforms), the resulting data is automatically processed using the core facility’s pipeline servers. The resulting data are staged on centrally managed network attached storage, and are subsequently processed to yield a standardized, compact data archive. This archive is communicated back to the requestor via web interfaces and made available for analysis using our local Galaxy server. Our system leverages open source tools to report quality control statistics that may uncover issues with either library or assay quality. These standardized data and QC results are made available via web interfaces to users and are easily sharable with both their collaborators and entire lab groups. Further, our flexible data model facilitates the annotation of relationships between the sample and assays. The end result is that the researcher and their collaborators can visualize the overall experimental design, and are able to access all the relevant data within the study.
Database accounts are given to only:
Princeton University Central Authentication Service handles the project's login credentials. Provided you can already authenticate here, and you are one of the above, you may register for an database account here. Typically, accounts for the Core's customers are usually sponsored and created by the Core-staff, without the need for independent registration [i.e. they fill and submit the form, for you]. If you are a Princeton researcher, and need to sponsor an external collaborator, please consult the Guest Account Provisioning (GAP) service.
Authentication is currently handled through Princeton's CAS. The curators do not know your password, so we can't remind you. Passwords must be retrieved changed through Princeton processes, clicking the "Forgot your password?" link when given the opportunity.
Transparently recording experimental provenance is important to us. Sequencing studies typically involve dozens of samples which are pooled/multiplexed, sequenced multiple times, and the resulting data processed in a myriad of ways. We flexibly model samples, assays, and datasets as nodes within a directed acyclic graph. Graph edges are the protocols documenting the procedures and data transformations between the nodes. For more detail, please see, "What is the experimental graph?"
The database can grant access to any known contact, be they individual or group. There is always a balance between pre-publication data confidentality and the facilitation of collaborative research with your peers. By default, the database associates each user account within a "primary" lab group. This group is granted view access to any samples (and their outputs) owned by that user, automatically. Therefore, everyone within a research lab should be able to view each other's samples and derived data.
Further, if an investigator is looking at a record, it makes sense that they can see everything that went into it (aim: transparent experimental provenance). Generally speaking within a scientific workflow, any child records will inherit the parent's privileges when that record is produced, by default. Therefore, a new assay is always created with the union of privileges from the combined input sample(s). Adding privileges after the fact, though, is handled slightly differently. To support transparent experimental provenance, granting view access to a record will also propagate view access to all upstream/ancestor records within that experiment graph. We do not do the same for downstream/children derivative records, because of issues with modeling source-samples (common laboratory strains, etc.). Therefore, it is recommended that you grant collaborators access to the most downstream assay or dataset, if you wish them to see the entire workflow. For more discussion (including the converse action, removing access), please consult : "Who can see my data?".
The database stores both nucleic acid preparations and constructed libraries. It tracks the sample's organism, the design intent (RNAseq, ChIPseq, etc.), protocol and crucial parameters, quality control details, tube identifier, concentration, and other important attributes. Generally, our specifications follow our genomics core's guidelines.
NOTE: LSI computing and research data storage services are not intended or appropriate for human subjects, sensitive data, or data covered by user agreements that require specific security or privacy controls. Please contact LSI computing support (csgenome@princeton.edu) for assistance or guidance on resources that may be appropriate for this or other restricted data.
As of July 1, 2016, all samples and sequencing requests are typically entered through the Princeton University Core Facilities iLab Portal. The Core staff will transmit your samples and requests into the database.
For those unrestricted researchers that construct their own complex libraries, and wish to track all the layers of sample provenance (sample protocols, sample multiplexing, library constructs, etc.), there are two sample submission interfaces:
Barcode files are associated with samples and are usually created by the Core when they construct libraries. They are used after sequencing to demultiplex the resulting data. A sample can have two barcode files associated with it: an "official" and a "custom". The official barcode file is generated automatically based on the barcodes for each sample. A custom barcode file can also be constructed as need warrants. If you submit already pooled libraries you can upload a custom barcode file to accompany them. The structure of a barcode file is simple, containing two or three tab-delimited columns. The first column is the sample name, the second is the barcode sequence. If a dual-indexing strategy is employed the third column contains the second barcode. Lines beginning with "#" are ignored.
# Name Index1 Index2
sample1 ACTGACTG GTCACAGG
sample2 GACGACTA GTCACAGG
If you are submitting one or more pooled libraries into iLab, you can fill out a samplesheet to go along with your submission. The sheet will describe the samples that you pooled into your library(s) and include their barcodes. This will allow HTSEQ to create the constituent samples along with your pool. The format is very specific. It is similar to a barcode file but contains more information. Here is a samplesheet for the submission of two pools with 3 constituent samples each.
# Pool ID Sample ID Description Index1 (i7) Index2 (i5)
LCK01_19 LCK_01_19_1h 1/19 Timecourse 1h AGTCCACC AACAATCC
LCK01_19 LCK_01_19_2h 1/19 Timecourse 2h TGGCTACA AACAATCC
LCK01_19 LCK_01_19_3h 1/19 Timecourse 3h CGGATACC AACAATCC
LCK01_20 LCK_01_20_1h 1/20 Timecourse 1h GAGTTACG AACAATCC
LCK01_20 LCK_01_20_2h 1/20 Timecourse 2h AGTCTACA AACAATCC
LCK01_20 LCK_01_20_3h 1/20 Timecourse 3h GGGTTACT AACAATCC
Sample names must be unique in the database so please give them unique and descriptive names, e.g. avoid names like "Pool1", "Pool2". The pool name must be included even if you are submitting 1 sample. The pool names must match the sample names that you enter in iLab. Within a pool, the length of the barcodes must be the same.
An assay contains all the raw data from a sequencing run. This includes fastq files and fastqc statistics and reports. Assay data is archived indefinitely.
A dataset contains the derived data from a set of assays. There are two main types of datasets: merged and demultiplexed. For example, if a sample is sequenced on two lanes, two assays are produced and they are automatically merged by combining the fastq data from each read. If the sample(s) are barcoded, then the data is automatically demultiplexed and fastq files are generated for each sample. Datasets can be deleted and regenerated without touching the underlying raw assay data. HTSEQ provides interfaces to enqueue both merge and demultiplexing jobs.
There are two other types of datasets which are generated for 10X samples which use the Chromium™ protocol. Datasets can be run through cellranger count which generates a set of files including normalized matrices and analysis statistics. Those datasets can in turn be run through cellranger aggregate. For more information consult 10x Genomics' Cell Ranger Documentation, or jump to :"How do I run Cell Ranger on my data?".
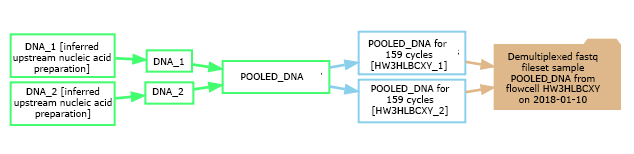
The experimental graph relates samples, assays and datasets and describes the workflow of the experiment. In the database's graphical representations, sample nodes are green, assay nodes are blue, and dataset nodes are tan. In the example shown here, two barcoded samples were pooled into a library, and that library was subsequently sequenced on two lanes of a flowcell, yielding two distinct assays. These two replicate assays were then merged and demultiplexed into a dataset containing demultiplexed fastq files and statistics for each sample. The graphs are clickable, with each node linking to a detailed view for that sample, assay, or dataset. The arrows (edges) are also clickable, linking to the experimental protocols describing the procedure or transformation between entities (if they were documented). You can view all of your experimental graphs on one page by clicking on "My Data" -> "Display My Graphs".
After your sample(s) are sequenced, the raw data from the sequencer are loaded as database assays with their associated fastq files and fastqc reports. If the sample(s) were run on multiple lanes of the same flowcell, the resulting assays are automatically merged, and if the samples are barcoded the data will then be demultiplexed. When the process is complete, you will receive an email with a link to your data.
The database uses a system of permissions (not unlike file system permissions) to protect your data and allow it to be viewed by others. When samples are transferred from iLab to the database, you are made the "owner" of them, which grants you "view" and "edit" privileges. That ownership is transferred to the assays and datasets which are created further down the pipeline. The other people who can see your data are the members of your lab. They have "view" access but not "edit" access. If you want other users of the database to see your data you can grant them access by going to page containing the sample/assay/dataset, pressing the "Edit" button at the top of the page and then going to the "Access" section which contains lists of individuals and labs. This is where you grant them "view" access. Note that if you grant access to a sample/assay/dataset to a user, that access is proprogated backwards in the chain of objects described in the experimental graph. For example, if you grant access to an assay, you are implicitly granting access to all the samples before it, but not to any datasets after it. Removing access works in the other direction: if you remove access to an assay, you are implicitly removing access to any datasets that are derived from it, but not from the samples that feed into it.
At times, you may need to authorize another researcher to update certain records. For instance, if a collaborator has provided you with a pre-made pooled library for sequencing and possesses expert knowledge of the index sequences of those records, you can permit them to update the barcodes for those samples/assays. To do so, access the edit interface (as described above), and click the “Add Owner” button to grant the collaborator additional privileges to that experimental graph.
There are a variety of ways to search for your data:
All searches are limited to the data which you have permission to view: your own data, the data
from other members of your lab,
and any other data to which you have specifically been given access.
Fastq files are associated with assays and datasets and links to them appear on the object's main page. You can click on individual links to download individual files. If you want to download a set of files, then press the "Download" button which appears wherever there is data to download. You will then be presented with lists of all the fastq files, partitioned by assay and dataset, formatted as commands for "wget" and "curl" (and also as plain links). Highlight the commands you want and copy them onto your clipboard. Paste the commands onto the command line of your target computer.
There are two ways to upload data to Galaxy. It can be done in batch by pressing the "Upload to Galaxy" button, which brings you to the same interface as used for downloading your data. There you will find a button to upload fastq files to Galaxy. You can also go from Galaxy to the Assay Search page, retrieve the specific assay and press the "Upload to Galaxy" buttons next to each fastq file.
The simplest way to share your data is to email them the wget/curl commands from the Batch Data Download page. Your collaborators do not need a database account. However, for those long-term collaborations, where the external researcher requires access to all your data, it may be preferable to sponsor them with a guest account. If so, please consult the Guest Account Provisioning (GAP) service.
All these accounts are distinct, but have some shared elements so that we can link up between services. In order to support Princeton CAS authentication, your database userid is the same as your Princeton NETID. In order to associate database sample-owners with iLab sample-submitters, it is best for the email addresses to be equivalent between these two accounts. To reduce ambiguity, ideally all three accounts should have the same email address.
The tube identifer should match what is physically written on the tube. Preferably, this is a unique barcode or ID that distinguishes it from any other tube that the core might handle [i.e. truly unique, and hopefully immutable]. You enter it under the "Sample ID" on the iLab form. Initially the sample names and tube identifiers are the same, as that is what the Core staff enters into the database. After entry, you can update the sample name to be more descriptive. The sample name is part of the fastq filename (see next question).
The fastq download file names are intended to convey the complete metadata for each file. Assay fastq files contain the sample name, flowcell and read number. Dataset fastq files contain the dataset type (i.e., merged, demultiplexed), the assay name, flowcell, run date and read number. If the sample or assay name is particularly long, you can edit it to make it shorter and the filename will change accordingly. You can of course rename your files after you have downloaded them to your computer.
You cannot delete samples or assays that have been loaded into HTSEQ. You can, however, delete your datasets, since they contain data derived from the raw assay data. For example, demultiplexing allows for 1 mismatch per index read by default. If you prefer to specify 0 or 2 mismatches, you might want to delete the demultiplexed dataset and submit a new demultiplexing job with the altered parameter. (Note that you do not have to delete the dataset in order to create a new one, provided that the new one uses different computing parameters.
To run Cell Ranger Count, go to the page of one of your demultiplexed datasets. In the "Derived Data" section there will be a "Cell Ranger Count" button. Click on that and then select the appropriate samples along with their reference genome. A job will be submitted for each sample and you'll receive an email when each is complete. To run Cell Ranger Aggregate go to the "Viewable Datasets" page (from "My Data" in the menubar) and select the cell ranger count datasets that you want to aggregate. You will be prompted for a name, description and normalization method. You'll receive an email when the job is complete.
The schema definition is available from the database table specifications page.
For more of the hardware and software platform, see this page.